Cory:
Unlock Your AI Assistant Now!
Abstract
The current standard regimen for antithrombotic therapy after percutaneous left atrial appendage closure (LAAC) in patients with non-valvular atrial fibrillation (NVAF) recommends long-term use of antiplatelet agents. However, this recommendation is not supported by sufficient clinical evidence. Since LAAC is a treatment option for managing patients at high risk of bleeding, it is necessary to clarify whether long-term antiplatelet therapy is truly required after LAAC. The Non-Antithrombotic Versus. Single antiPlatelet Therapy Following Left Atrial Appendage Closure (NAPT-LAAC) trial, a prospective, randomised, controlled, open-label, blinded-endpoint multicentre study, will be conducted in Japan. It was designed to evaluate whether non-antithrombotic therapy is non-inferior to antiplatelet monotherapy after 45 days of oral anticoagulant (OAC) monotherapy following LAAC, with respect to the incidence of thrombotic and bleeding composite events in patients with NVAF and high bleeding risk. Patients with NVAF with a CHA2DS2-VA score ≥2 and who successfully undergo LAAC are eligible for inclusion. A total of 500 patients undergoing LAAC will be randomised (1:1) to aspirin monotherapy versus non-antithrombotic therapy for the 45 days following OAC monotherapy. The primary outcome is a composite of all-cause mortality, myocardial infarction, stroke, systemic embolism, major bleeding, and clinically relevant non-fatal bleeding during a maximum of 4 years of follow-up. Major bleeding or clinically relevant non-fatal bleeding is defined as Type 2, 3, or 5 bleeding, according to the Bleeding Academic Research Consortium definition. The NAPT-LAAC trial will determine the probable non-inferiority of long-term non-antithrombotic therapy to aspirin monotherapy in patients with NVAF who undergo LAAC. (ClinicalTrials.gov: NCT07125417; jRCTs031250110)
In patients with non-valvular atrial fibrillation (NVAF), preventing thromboembolic disease, primarily due to left atrial appendage thrombi1, is one of the most important therapeutic goals. Currently, oral anticoagulant (OAC) therapy is the first-line treatment for preventing cardiogenic embolism caused by NVAF234. However, some patients do not tolerate long-term anticoagulation therapy owing to a high risk of bleeding; therefore, non-pharmacological treatment options to reduce the risk of embolism in these patients are required.
A percutaneous catheter-based left atrial appendage closure (LAAC) device has recently been developed and is expected to become an alternative therapeutic option to OAC for preventing cardiogenic embolism in patients with NVAF. The current standard antithrombotic regimen after LAAC comprises an initial 45-day course of OAC combined with single antiplatelet therapy, followed by dual antiplatelet therapy for up to 6 months, after which patients are transitioned to single antiplatelet therapy, with aspirin as the recommended agent. This antithrombotic regimen is designed to prevent ischaemic stroke and device-related thrombosis (DRT) after the procedure. However, this conventional drug regimen is currently used despite a lack of sufficient clinical evidence. Since the LAAC device was originally intended to be an alternative treatment option for managing patients at high risk of bleeding due to anticoagulant use, the current regimen should be updated.
We designed a clinical trial to evaluate whether non-antithrombotic therapy is non-inferior to antiplatelet monotherapy after 45 days of OAC monotherapy post-LAAC with respect to the incidence of thrombotic and bleeding composite events in patients with NVAF and high bleeding risk.
Methods
Trial structure and oversight
The Non-Antithrombotic Versus. Single antiPlatelet Therapy Following Left Atrial Appendage Closure (NAPT-LAAC) trial is a prospective, randomised, controlled, open-label, blinded-endpoint, multicentre trial being conducted in Japan to test the non-inferiority of non-antithrombotic therapy compared with aspirin monotherapy in patients who undergo percutaneous LAAC without indications for long-term OAC. The trial is being conducted in collaboration with the Optimized CathEter vAlvular iNtervention Left Atrial Appendage Closure (OCEAN-LAAC) registry5, as part of the OCEAN Structural Heart Disease (OCEAN-SHD) organisation. The trial is funded by Boston Scientific Japan KK. The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the manuscript. The trial protocol has been registered with the Japan Registry of Clinical Trials (jRCTs031250110) and ClinicalTrials.gov (NCT07125417). The study is being conducted in accordance with the principles of the Declaration of Helsinki, Ethical Guidelines for Medical and Biological Research Involving Human Subjects, the Clinical Trials Act, and other current legal regulations in Japan. This study was approved by the Certified Review Board of Hattori Clinic, Tokyo, Japan (certified number: CRB3180027; review number: 81-A). Data are managed by third-party entities (EPS Corporation, Shinjuku-ku, Tokyo, Japan) using a computer-based data management system (EzCap [Hawkie MediTech]). The trial is monitored by an independent data and safety monitoring board and, based on their recommendations, decisions related to safety and trial continuation are made by the executive committee.
Study participants
Patients with NVAF who have a CHA2DS2-VA score ≥2 and who successfully undergo LAAC are eligible for inclusion. The indications for LAAC are determined by individual local Heart Team discussions, in accordance with the current guidelines in Japan6, which are substantially identical to those in the US and Europe27. Details of the inclusion and exclusion criteria are summarised in Table 1 and Table 2, respectively.
The LAAC procedure is performed using the WATCHMAN FLX Pro device (Boston Scientific) in all cases. The procedure is typically carried out in accordance with the manufacturer’s instructions. However, this does not prevent the physician at each participating hospital from selecting the optimal procedure for each patient.
A total of 500 patients undergoing LAAC will be randomised (1:1) to aspirin monotherapy (75-100 mg) or non-antithrombotic therapy at 20 Japanese centres. Patients will be recruited within 1 day after the LAAC procedure and randomised to either arm. All patients should provide written informed consent prior to study enrolment. The end of anticipated enrolment will be in May 2027.
Table 1. Inclusion criteria.
| Patients will be included if they meet all the following criteria. |
|---|
| 1) Patient has documented NVAF (i.e., AF without severe mitral stenosis or mechanical valves) |
| 2) Patient has a CHA2DS2-VA score of 2 or greater |
| 3) Patient meets the guidelines for proper use of the LAAC system below, including patients who have an increased risk of bleeding*. Patients with high bleeding risk, including 1 or more of the following: a. Subject with a HAS-BLED score of 3 or higher b. Patient with multiple previous injuries associated with falls who required treatment c. Patient with a history of diffuse cerebral amyloid angiopathy d. Patient requiring long-term (>1 year) concomitant use of 2 or more antiplatelet drugs e. Patient with a history of major bleeding classified as Type 3 by the Bleeding Academic Research Consortium |
| 4) Individual with NVAF who has undergone successful LAAC (defined as no significant residual circumferential leak [>3 mm] or major morbidity by the time of procedure completion) |
| 5) Patient is suitable for pharmacotherapy as defined in this study protocol in both the NAPT and SAPT arms |
| 6) LAA anatomy is suitable for WATCHMAN FLX Pro implantation |
| 7) LAA anatomy is suitable for an LAAC procedure |
| 8) The patient and the investigator and/or subinvestigator agree that the patient will return for all required visits after the LAAC procedure |
| 9) Patient has thoroughly understood the purpose of the study and has provided informed written consent to participate in the study |
| *Selection criteria from6. AF: atrial fibrillation; LAA: left atrial appendage; LAAC: left atrial appendage closure; NAPT: no antithrombotic therapy; NVAF: non-valvular atrial fibrillation; SAPT: single antiplatelet therapy |
Table 2. Exclusion criteria.
| Patients who meet any of the following conditions will be excluded. |
|---|
| 1) Patients who are currently enrolled in other clinical trials, except when the patient is participating in mandatory governmental registries or purely observational registries with no associated treatment |
| 2) Individuals requiring long-term anticoagulation therapy for reasons other than AF-related stroke risk reduction (e.g., thrombophilic conditions, previous pulmonary embolism, or deep venous thrombosis) |
| 3) Patients requiring oral antiplatelet therapy for reasons other than LAAC (e.g., history of myocardial infarction, history of PCI, history of endovascular treatment, history of stroke/transient ischaemic attack, significant coronary stenosis proven by myocardial ischaemia, severe carotid stenosis requiring invasive treatment, or if the investigator and/or subinvestigator judged the need for antiplatelet therapy) |
| 4) Patients who meet 1 or more of the following criteria: a. Patients who are contraindicated for DOAC or VKA b. Patients with a contraindication to aspirin c. Patients diagnosed with an allergy to aspirin |
| 5) Patients who have undergone or are scheduled to undergo cardiac or non-cardiac intervention or surgery within 45 days before or 60 days after LAAC (e.g., cardioversion, PCI, cardiac ablation, cataract surgery, other structural heart interventions) |
| 6) Patients with stroke (either ischaemic or haemorrhagic) or transient ischaemic attack within 30 days prior to enrolment |
| 7) Patients with active bleeding |
| 8) Patients who lack LAA or whose LAA has been surgically ligated |
| 9) Patients who experienced a myocardial infarction (with or without intervention) recorded as a non-ST-segment elevation myocardial infarction or ST-segment elevation myocardial infarction in the 30-day period prior to enrolment |
| 10) Patients with a previous atrial septal repair or with an atrial septal defect/patent foramen ovale device |
| 11) Patients with mechanical valve prostheses at any site |
| 12) Persons with known contraindications to TOE |
| 13) Patients with active infection |
| 14) Individuals with NYHA Class IV congestive heart failure at enrolment |
| 15) Patients who are pregnant, breastfeeding, or wishing to become pregnant |
| 16) Patients with a life expectancy of less than 2 years |
| 17) Patients requiring emergency surgery for any reason |
| 18) Patients who, at the discretion of the investigator, have other medical, social, or psychological conditions that preclude adherence to appropriate consent or the follow-up tests required by the protocol |
| 19) Patients whose investigator or subinvestigator judges their participation in the study to be inappropriate |
| AF: atrial fibrillation; DOAC: direct oral anticoagulant; LAA: left atrial appendage; LAAC: left atrial appendage closure; NYHA: New York Heart Association; PCI: percutaneous coronary intervention; TOE: transoesophageal echocardiography; VKA: vitamin K antagonist |
Study procedures
Randomisation and treatment allocation
Once a patient fulfils the criteria for randomisation and provides written informed consent, investigators will use an electronic data capture system (EzCap) to obtain the treatment assignment. Randomisation (1:1) will be performed using a permuted block design to ensure an approximate balance between the treatment groups (aspirin monotherapy versus non-antithrombotic therapy) via a computer pseudo-random number generator. Randomisation will be stratified by institute, OAC type, CHA2DS2-VA score, and HAS-BLED score. In both groups, OAC will be administered before the LAAC procedure and terminated 45-59 days after the procedure. The choice of the type of OAC will be left to the discretion of the treating physician to ensure optimal patient care. In the aspirin monotherapy group, aspirin administration will be initiated immediately after terminating OAC therapy. In the non-antithrombotic drug group, antithrombotic drugs will not be administered after terminating OAC therapy.
Follow-up
The flowchart of this study is shown in Figure 1. Enrolled patients will be followed up for a maximum of 4 years or until study completion (31 May 2029). Follow-up visits for at least 2 years after enrolment will be mandatory (3 timepoints: 45 days [visit 1], 1 year [visit 2], and 2 years [visit 3]), with additional visits (end of the observational period [visit 4]) scheduled according to the enrolment period. At each follow-up visit, compliance with the investigational drug, concomitant medical therapy, assessment of cardiovascular outcomes, and adverse events will be checked (Figure 2). After identifying a potential event, the local investigator will collect and send the relevant information to the clinical trial coordination unit for blinded event adjudication by an independent clinical events committee. Participants who require a treatment different from their randomised assignment during the observation period and consequently discontinue the assigned study treatment will be considered as having discontinued the study treatment.
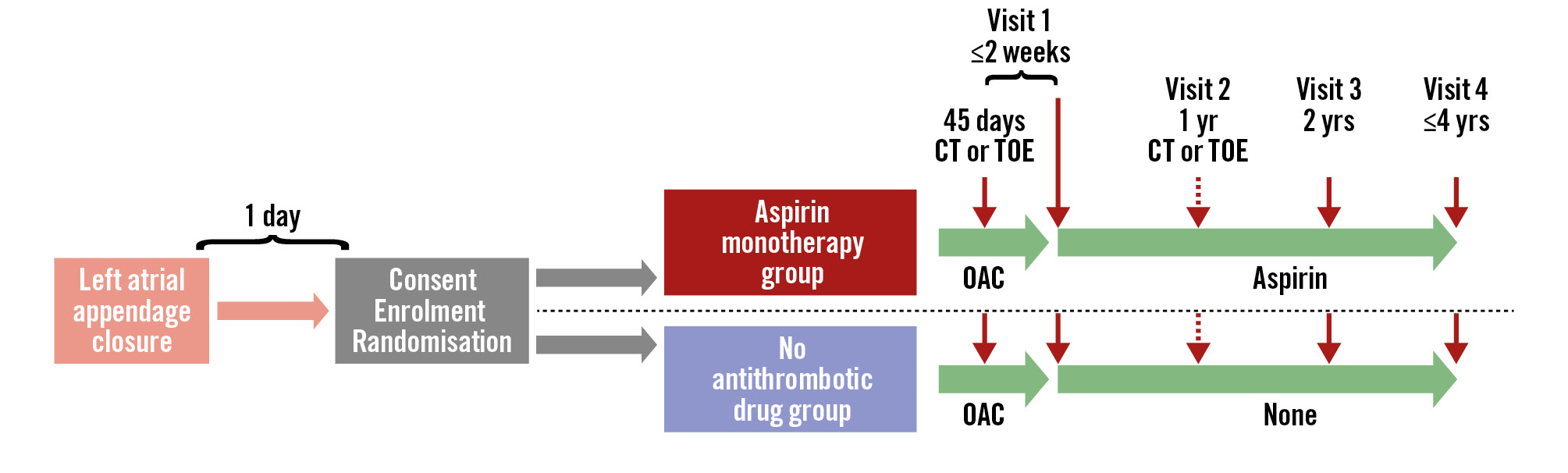
Figure 1. Flowchart of the study design. CT: computed tomography; OAC: oral anticoagulant; TOE: transoesophageal echocardiography
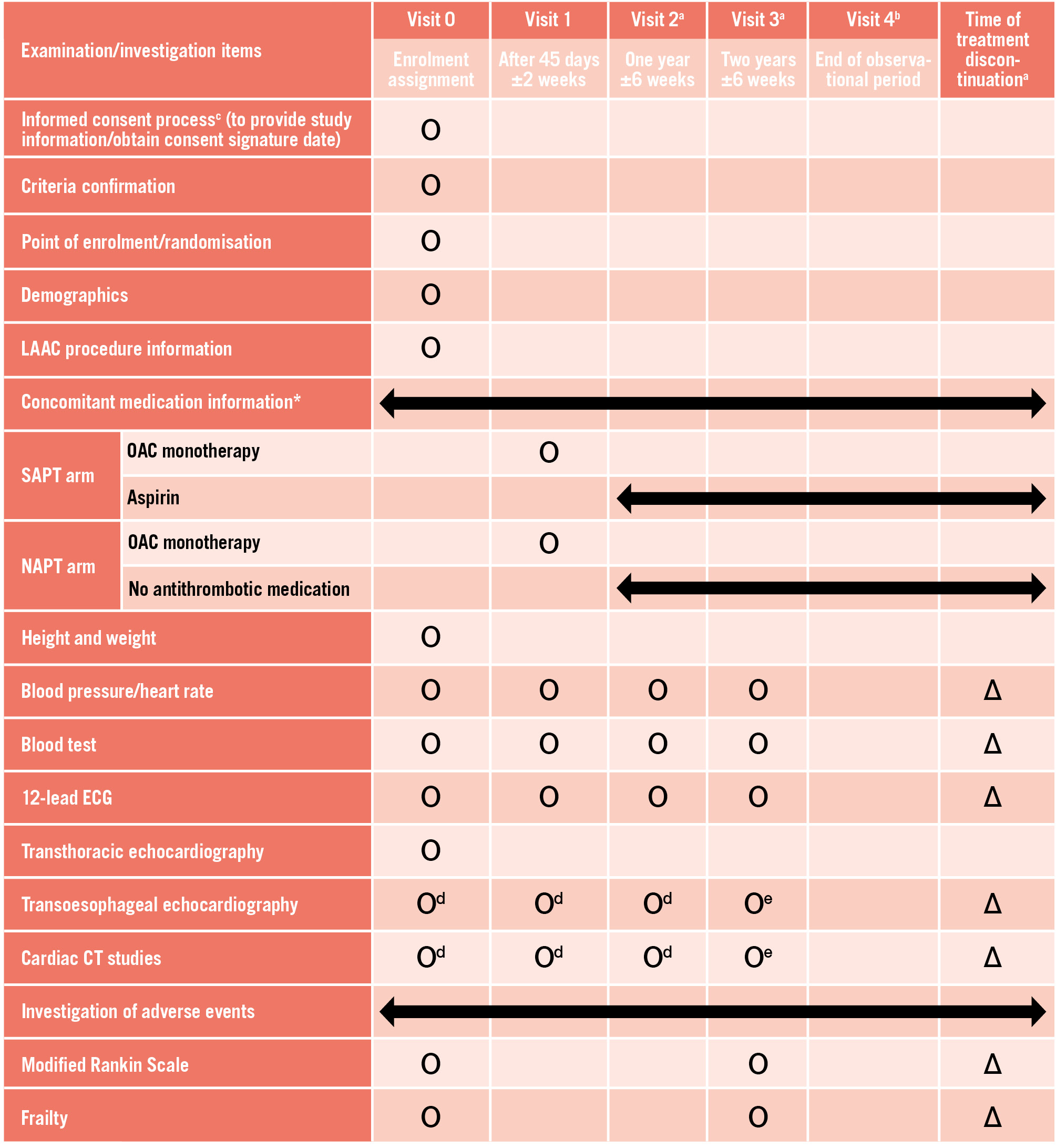
Figure 2. Study schedule. Ο Indicates scheduled assessments. ∆ Indicates assessments performed as clinically indicated. aTelephone interviews may be used to collect information on clinical status, concomitant medications, and adverse events when a participant is unable to return for follow-up due to relocation or transfer to another hospital. bThe observational period extends up to 4 years after enrolment of the first patient and applies to participants followed for more than 2 years and 6 weeks after visit 3. Telephone interviews are the standard method of follow-up; however, routine office visits may also be used for observational data collection. cProviding study information prior to the LAAC procedure is permitted; however, written informed consent must be obtained on the day following the procedure. dTOE and/or cardiac CT will be performed according to the study schedule. eTOE or cardiac CT will be performed at the physician’s discretion; however, in cases of suspected embolic events, assessment must be performed using TOE or cardiac CT. *In addition to the clinical interview, antithrombotic medication adherence will be assessed by reviewing the patient’s medication diary at each office visit. CT: computed tomography; ECG: electrocardiography; LAAC: left atrial appendage closure; OAC: oral anticoagulant; TOE: transoesophageal echocardiography
Study outcomes
The primary and secondary outcomes are listed in Table 3. In brief, the primary outcome is a composite of all-cause mortality, myocardial infarction, stroke, systemic embolism, major bleeding, or clinically relevant non-fatal bleeding from randomisation to the end of the study observation period (up to a maximum follow-up of 4 years). For the secondary outcome, when DRT is detected on computed tomography and/or transoesophageal echocardiography, appropriate OAC therapy and follow-up imaging will be performed at the discretion of the treating physician, and patients will be subsequently followed according to the intention-to-treat principle. Surviving cases of no events will be censored as of the date of discontinuation of the study treatment or the date of the last confirmation of survival. The explanatory outcome is listed in Table 4.
Table 3. Primary and secondary endpoints.
| Primary endpoint |
|---|
| A composite endpoint consisting of all-cause mortality, myocardial infarction, stroke, systemic embolism, and major/clinically relevant non-fatal bleeding* from randomisation to the end of study observation (up to a maximum follow-up of 4 years) |
| Secondary endpoint |
| 1) All procedure-unrelated† BARC Type 1 bleeding or greater from randomisation to the end of follow-up‡ |
| 2) ISTH major bleeding unrelated to the procedure and clinically relevant non-fatal bleeding† from randomisation to the end of follow-up‡ |
| 3) Incidence of DRT based on CT and/or TOE at visit 1, visit 2 (1 year), and visit 3 (2 years) after randomisation |
| 4) A composite endpoint consisting of the occurrence of ischaemic stroke and systemic embolism from randomisation to the end of follow-up‡ |
| *Major/clinically significant non-fatal bleeding is defined as Type 2, 3, or 5 bleeding among the BARC bleeding definitions. †Procedure-unrelated bleeding is defined as occurring ≥7 days after the LAAC procedure. ‡These outcomes will also be collected from the day following visit 1 to the end of follow-up. BARC: Bleeding Academic Research Consortium; CT: computed tomography; DRT: device-related thrombosis; ISTH: International Society on Thrombosis and Haemostasis; LAAC: left atrial appendage closure; TOE: transoesophageal echocardiography |
Table 4. Explanatory endpoints.
| 1) Incidence of the following early safety composite endpoints from randomisation to visit 1: a. Overall mortality b. Ischaemic stroke (with/without occlusion; with/without disability) c. Systemic embolism, excluding cerebral infarction d. Myocardial infarction e. All bleeding f. Acute kidney injury stage 2 or 3 (including renal replacement therapy) g. Readmissions related to procedure-related complications (vascular access site, procedure-related bleeding, device embolisation, pericardial effusion requiring intervention) h. Unexpected procedure-related hospitalisation i. Patient adherence to OAC monotherapy |
| 2) Incidence of the following clinical endpoints during the period from randomisation to the end of study observation (4-year period at the maximum): a. Cardiovascular death b. Ischaemic stroke (with/without occlusion; with/without disability) c. Haemorrhagic stroke rates d. Systemic embolism, excluding cerebral infarction e. Myocardial infarction f. Major bleeding rates g. All bleeding h. Unexpected hospitalisation i. Patient adherence rates to OAC monotherapy j. Patient adherence to aspirin k. Discontinuation of antithrombotic medications |
| 3) Survival time analyses for the primary and secondary endpoints, censored at the date of the event in cases where the event occurred, and at the date of the last survival check in cases where the event did not occur, regardless of discontinuation of study treatment |
| OAC: oral anticoagulant |
Statistical considerations
Sample size calculation
In the Japanese Primary Prevention Project (JPPP) study on primary prevention with aspirin in Japanese individuals, the 5-year cumulative incidence of fatal and non-fatal cardiovascular events and non-cardiovascular death was not statistically significant, with an overall rate of 7.71%8, while the 5-year cumulative incidence of major haemorrhage was 0.86% and 0.51% in the aspirin and control groups, respectively. According to unpublished interim data from the Non-antithrombotic Therapy After Transcatheter Aortic Valve Implantation Trial (currently being conducted by the OCEAN-SHD Study Group)9, minor bleeding was approximately 3 times more common than major bleeding. Therefore, the 5-year cumulative incidence of all bleeding events in the JPPP study was estimated to be 3.44% and 2.04% in the aspirin and control groups, respectively. Considering these data, the 5-year cumulative incidence rate for the primary endpoint of the NAPT-LAAC trial is estimated to be 11.15% (7.71%+3.44%) and 9.75% (7.71%+2.04%) in the aspirin and control groups, respectively, resulting in event-free rates of 88.85% and 90.28%, respectively, with a hazard ratio of 0.868 (control group vs aspirin group).
According to unpublished data from the ongoing OCEAN-LAAC registry, the 1-year cumulative event-free rate of the primary endpoint among patients receiving antiplatelet monotherapy is 87.7%. Therefore, by multiplying the rate by a hazard ratio of 0.868, the 1-year cumulative event-free rate in the non-antithrombotic therapy group is estimated to be 89.2%.
The non-inferiority margin in this trial is set at Δ5%, representing a clinically acceptable range. Based on a 1-year cumulative event-free rate of 87.7% in the antiplatelet monotherapy group, this corresponds to a non-inferiority threshold of 82.7%. The appropriateness of this margin was determined through consensus discussions among LAAC treatment experts affiliated with the OCEAN-SHD Study Group.
Based on these considerations, the required number of events and patients is calculated as 140 and 226, respectively, with a significance level of α=0.025 (1-sided test), β=0.15 (power 0.85), 2 years of enrolment, and a maximum follow-up of 4 years. To account for a 10% dropout rate, the final enrolment is set at 250 patients per group, totalling 500.
Statistical analysis
Primary and secondary outcomes will be evaluated in the full analysis set on an intention-to-treat basis. Outcomes will be collected until study completion regardless of treatment discontinuation. A per-protocol analysis may also be performed, as appropriate, for sensitivity analyses. If non-inferiority is demonstrated for the primary endpoint and the cumulative incidence is lower in the non-antithrombotic group than in the aspirin monotherapy group, superiority testing will be conducted using a 2-sided significance level of α=0.05. Safety analyses will be performed in the safety population. All analyses will be conducted after study completion and data lock. A separate statistical analysis plan will provide detailed descriptions of the statistical methods, including procedures for data handling.
Survival time analysis (Kaplan-Meier curve estimation and Cox proportional hazards model) will be performed for the primary outcome from randomisation to the end of the study observation period (up to a maximum follow-up of 4 years). Subgroup analysis is planned to be conducted based on the content shown in Supplementary Table 1. For secondary outcomes, survival-time analyses will also be performed. Depending on the number of events, a chi-squared test, which does not consider time-to-event in terms of cumulative incidence, will be performed. The same calculation will be performed from the day after visit 1 until the end of the study period. For exploratory outcomes, the incidence of DRT as examined with computed tomography or transoesophageal echocardiography will be assessed at 1 year after LAAC and compared using the chi-squared test. Additional exploratory assessments will include survival time analyses for the primary and secondary endpoints, censored at the date of the event, in cases wherein the event occurs, or at the date of the last survival check, in cases wherein the event does not occur, regardless of study treatment discontinuation.
Discussion
This study is designed to evaluate the non-inferiority of withholding long-term antithrombotic therapy after LAAC in patients with NVAF who are at high risk of bleeding. The primary purpose of administering antiplatelet agents after LAAC is the prevention of thromboembolic events, including DRT. However, there is currently no clear clinical evidence supporting the long-term use of antiplatelet agents after LAAC. Thus, recommendations are largely based on the empirical adaptation of the drug regimen used in clinical trials to develop LAAC devices. In this regard, the results of the present trial may provide evidence that prolonged antiplatelet therapy is unnecessary in patients with NVAF and high bleeding risk after LAAC.
Rodés-Cabau et al reported that prothrombin fragment 1+2 and thrombin-antithrombin III levels − biomarkers of thrombin generation − increased 7 days after the LAAC procedure and decreased thereafter10. In contrast, markers of platelet activation − soluble P-selectin and soluble CD40 ligand levels − did not increase at any time after the LAAC procedure10. These observations demonstrate significant activation of the coagulation system soon after the LAAC procedure without platelet activation. A recent clinical study has also shown that antiplatelet therapy does not attenuate coagulation system activation for up to 6 months after LAAC11. Taken together, these findings suggest that antiplatelet agents may play only a minimal role in preventing thromboembolic events after LAAC. Furthermore, withholding antiplatelet therapy after LAAC may reduce the risk of bleeding associated with these agents. Antiplatelet therapy is well known to show beneficial effects in the secondary but not primary prevention of atherosclerotic cardiovascular disease812. The protocol of the present study excludes patients with significant coronary and/or carotid artery stenosis and those with any history of atherosclerotic cardiovascular disease. This makes predicting the preventive effect of antiplatelet therapy against atherosclerotic cardiovascular disease in the present study difficult. From this perspective, this study may provide evidence that antiplatelet therapy is unnecessary after LAAC.
A recent meta-analysis has reported that OAC use for the initial 45 days after LAAC, compared with that for the initial 3 months, significantly reduces the risk of major bleeding without compromising the preventive effects against stroke, transient ischaemic attacks, and DRT13. One of the main mechanisms causing DRT to occur in the early phase after LAAC is direct contact of its metal frame and the fabric (e.g., expanded polytetrafluoroethylene or polyethylene terephthalate) with blood before endothelialisation of the device surface14. Therefore, the current standard regimen of OAC use for the initial 45 days after LAAC, which is also adopted in the protocol of this study, is considered an evidence-based recommendation. However, concerns persist about the risk of thrombotic complications, including DRT, in the late phase after LAAC. In this respect, long-term (a median of 13 months), half-dose OAC therapy has been reported to lower both bleeding and thromboembolic events compared with standard antithrombotic therapy after LAAC (with WATCHMAN implantation)15. The present study will use the latest-generation LAAC device, the WATCHMAN FLX Pro implant, which has a fluoropolymer coating that is designed to reduce inflammation and promote faster and more complete endothelialisation. Animal pathological studies showed that the WATCHMAN FLX Pro implant reduced the occurrence of DRT and promoted mature endothelialisation compared with conventional devices without any antithrombotic medications16. These observations suggest that non-antithrombotic therapy could minimise bleeding without substantially increasing late thromboembolic events in patients who undergo WATCHMAN FLX Pro implantation. This hypothesis warrants formal investigation in this randomised controlled trial, particularly to confirm adequate endothelial coverage through serial transoesophageal echocardiography or cardiac computed tomography examinations.
A randomised clinical trial addressing a similar question, the ASPIRIN LAAO trial17, is currently underway in China. Compared with the ASPIRIN LAAO trial, the present study uses a shorter duration of postprocedural OAC therapy (45-59 days vs 6 months). In addition, the present study focuses on the latest-generation WATCHMAN FLX Pro device, whereas the ASPIRIN LAAO trial protocol does not specify the device generation. Overall, the present study reflects contemporary device technology and postprocedural management and is expected to provide incremental evidence beyond the ASPIRIN LAAO trial.
Limitations
This study has some limitations. First, the trial uses a randomised controlled, open-label blinded endpoint design, which may introduce bias, despite the blinded endpoint adjudication. In particular, because the choice of the type of OAC during the first 45-59 days after the procedure is left to the discretion of the treating physician, it cannot be completely excluded that the OAC regimens may differ between the two groups despite randomisation. Second, since the study is being conducted exclusively in Japanese centres, the enrolled population may be characterised by a smaller body size and a higher risk of major bleeding compared with Western cohorts. Third, although the sample size is sufficient for primary non-inferiority analysis, it may be insufficient to detect other clinically significant outcomes, such as DRT. Finally, as with any randomised trial, treatment adherence and crossover cannot be fully controlled for, which may influence the interpretation of long-term outcomes.
Conclusions
This trial will determine the non-inferiority of long-term non-antithrombotic therapy to aspirin monotherapy in patients with NVAF who undergo LAAC. The results of this trial will benefit patients with NVAF and high bleeding risk by eliminating the need for long-term antiplatelet therapy after LAAC.
Acknowledgements
We would like to thank Editage for English language editing.
Funding
The study is funded by Boston Scientific Japan K.K.
Conflict of interest statement
M. Asami reports receiving honoraria for proctoring, speaker fees, and consulting fees from Boston Scientific Japan K.K.; speaker fees and consulting fees from Abbott; and lecture fees from Daiichi Sankyo Co., Ltd., Boehringer Ingelheim Japan, Inc., and Canon Inc. D. Hachinohe reports receiving honoraria for proctoring from Boston Scientific Japan K.K. K. Hayashida reports receiving lecture fees and consulting fees from Daiichi Sankyo Co., Ltd. Y. Izumi reports receiving lecture fees from Bristol-Myers Squibb K.K. S. Kubo reports receiving honoraria for proctoring, speaker fees, and consulting fees from Boston Scientific Japan K.K. and Abbott Japan Co., Ltd. T. Morikawa reports receiving honoraria for proctoring and lecture fees from Boston Scientific Japan K.K. and Abbott Japan Co., Ltd. M. Nakashima reports receiving honoraria for proctoring and lecture fees from Boston Scientific Japan K.K.; and lecture fees from Abbott Japan Co., Ltd. G. Nakazawa reports receiving honoraria or consulting fees from Boston Scientific Japan K.K. and Daiichi Sankyo Co., Ltd. Y. Ohno reports receiving speaker fees from Boston Scientific Japan K.K. and Abbott Japan Co., Ltd. S. Okazaki reports receiving lecture fees from Abbott Japan Co., Ltd. T. Otsuka reports receiving research grants and funding from Daiichi Sankyo Co., Ltd., and Canon Inc. M. Saji reports receiving speaker fees from Boston Scientific Japan K.K.; and speaker fees and lecture fees from Daiichi Sankyo Co., Ltd. and Bristol-Myers Squibb K.K. T. Shimura reports receiving honoraria for proctor and lecture fees from Abbott Japan Co., Ltd.; and lecture fees from Boston Scientific Japan K.K. S. Shirai reports receiving lecture fees from Boston Scientific Japan K.K. and Bristol-Myers Squibb K.K. A. Sugiura reports receiving lecture fees from Daiichi Sankyo Co., Ltd. and Abbott Japan Co., Ltd. H. Ueno reports receiving honoraria for proctor and lecture fees from Boston Scientific Japan K.K. Y. Watanabe reports receiving lecture fees from Boston Scientific Japan K.K. and Daiichi Sankyo Co., Ltd. M. Yamamoto reports receiving honoraria for proctoring and lecture fees from Boston Scientific Japan K.K. and Abbott Japan Co., Ltd.; and lecture fees from Daiichi Sankyo Co., Ltd. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
