Cory:
Unlock Your AI Assistant Now!
Abstract
Bioprosthetic mitral valves are prone to structural valve deterioration (SVD) over time, which can lead to bioprosthetic valve dysfunction (BVD) requiring reintervention. Surgical redo-mitral valve replacement (rMVR) is currently the standard treatment, although it is associated with significant mortality in high-risk patients. Transcatheter mitral valve-in-valve (mViV) has emerged as an alternative to surgical rMVR in patients with failed bioprostheses, but randomised studies comparing the two treatments are lacking. The SURVIV trial is an investigator-initiated, prospective, multicentre, open-label, randomised controlled trial that will enrol 150 patients with mitral BVD suitable for surgical rMVR or transcatheter mViV. Participants will be randomised 1:1 to transseptal mViV with a balloon-expandable transcatheter heart valve or to conventional surgical rMVR. Procedures will be performed according to local best practices with contemporary medical devices. The primary endpoint is the composite of all-cause mortality or disabling stroke at 12 months. Key secondary endpoints are major complications (cardiovascular death, disabling stroke, life-threatening or major bleeding, acute kidney injury stage 2 or 3, and major vascular complications) at 30 days, according to Mitral Valve Academic Research Consortium criteria; rehospitalisation for cardiovascular causes at 12 months; echocardiographic and/or tomographic signs of prosthetic valve thrombosis and early SVD at 3 and 12 months; and health-related quality of life (EQ-5D-5L) at 3 and 12 months. Clinical follow-up will continue up to 10 years. SURVIV is the first randomised trial to compare a transcatheter mViV procedure with surgical rMVR for mitral BVD and may provide further clinical evidence to guide the management of patients with failed mitral bioprostheses. ClinicalTrials.gov: NCT04402931.
Bioprosthetic valves are the device of choice for the majority of patients who undergo surgical mitral valve replacement, and their use has grown steadily as clinicians and patients seek to avoid lifelong anticoagulation1. Although multiple cohort studies have shown excellent survival and freedom from valve-related complications, all bioprosthetic valves are prone to progressive tissue deterioration – leading ultimately to structural bioprosthetic valve dysfunction (BVD). Surgical redo-mitral valve replacement (rMVR) remains the established therapy for BVD2, but this can be associated with high mortality rates, particularly in patients with advanced disease, comorbidities, or multiple previous cardiac surgeries34567. Moreover, technical operative difficulties caused by tissue adhesions substantially increase the occurrence of bleeding complications, leading to significant morbidity and increased in-hospital length of stay.
Transcatheter valve-in-valve has emerged as a less invasive alternative to redo-surgery for older, high-risk patients with BVD. Initially applied for treating failed aortic bioprostheses8, transcatheter mitral valve-in-valve (mViV) replacement has now been adopted worldwide. Contemporary registries report low rates of periprocedural morbidity and mortality in high- and intermediate-risk patients, coupled with favourable valve performance91011. Recent meta-analyses comparing mViV and rMVR have shown equivalent mortality but lower incidences of stroke, major bleeding, and acute kidney injury with mViV12131415. Most available data are derived from observational studies subject to selection bias, heterogeneous follow-up, and incomplete adjustment for confounding161718. Therefore, a head-to-head comparison of the results of mViV and rMVR is crucial to guide clinical recommendations (especially in younger, lower-risk patients). Additionally, careful imaging follow-up is required to detect complications such as leaflet thrombosis, late transcatheter heart valve (THV) migration19 and structural valve deterioration (SVD); such data are sometimes difficult to obtain from non-controlled trials. The Randomized Trial of Transcatheter Valve-In-Valve vs Redo Surgery for Biosprosthetic Mitral Dysfunction (SURVIV) aims to compare the aforementioned treatments in patients across the surgical risk spectrum with mitral BVD. Applying standardised endpoint definitions and comprehensive echocardiographic and tomographic assessment, its primary aim is to determine whether transcatheter mViV surpasses surgical rMVR in safety and efficacy.
Methods
Rationale and study design
The SURVIV study is an investigator-initiated, prospective, randomised, multicentre, controlled, open-label clinical trial designed to assess the safety and efficacy of transseptal mViV as compared to repeat surgical mitral valve replacement in patients with severe mitral BVD. Seven participating centres from different regions in Brazil (4 in the southeast, 2 in the northeast, and 1 in the south) have been selected, all with expertise in both transcatheter and surgical mitral valve interventions. Each centre maintains a dedicated Heart Team comprising interventional cardiologists, cardiac surgeons, imaging specialists, and clinical coordinators to ensure optimal patient care and protocol adherence.
The trial will be conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Ethical approval has been obtained from the institutional review boards of all participating centres, and the study is registered at ClinicalTrials.gov: NCT04402931 (date of registration: 17 Feb 2020). All participants will provide written informed consent before enrolment. The study flow is depicted in Figure 1.
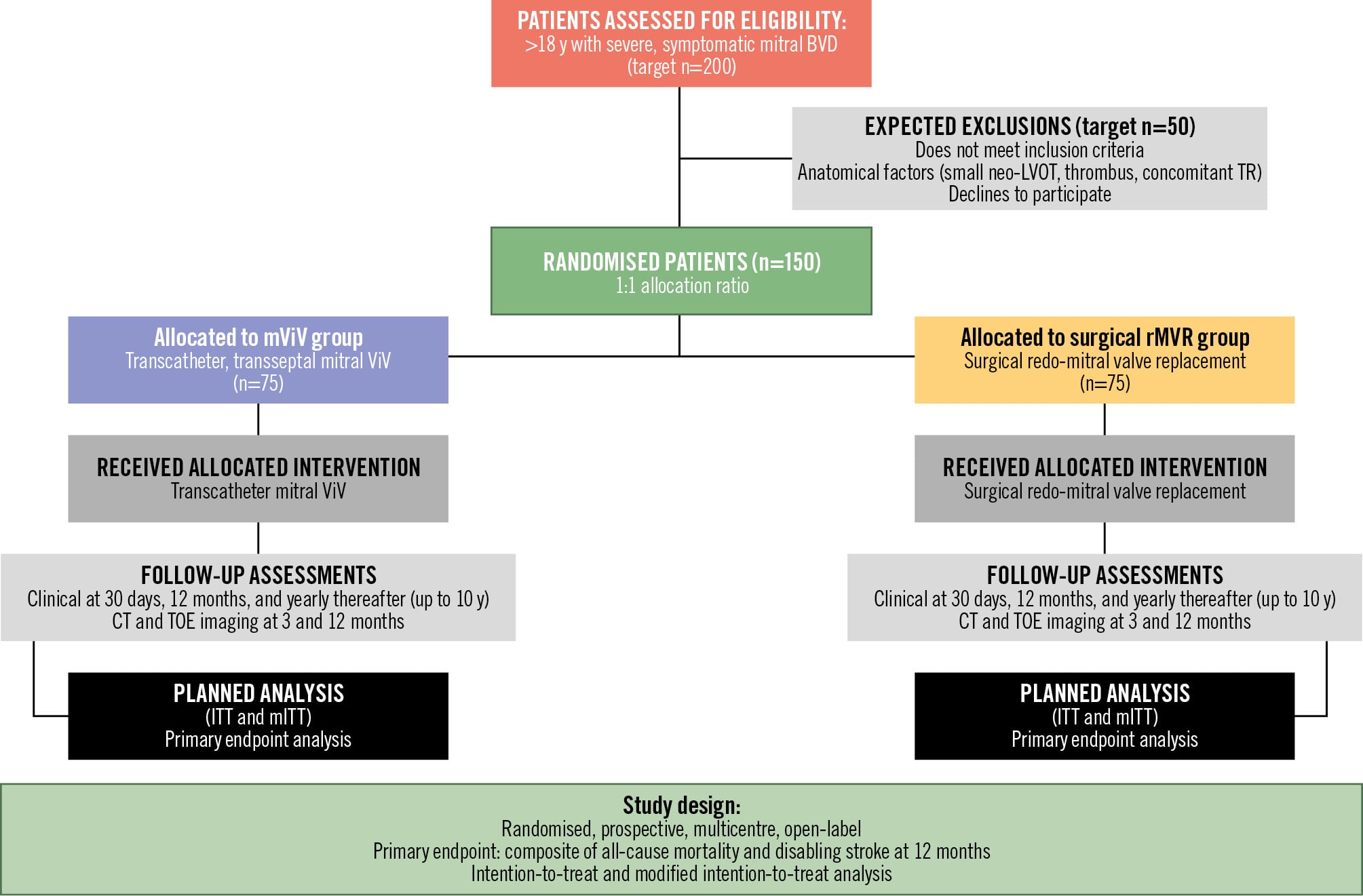
Figure 1. Planned study flow diagram for the SURVIV trial. This CONSORT flow diagram illustrates the planned recruitment, randomisation, intervention, and follow-up strategy for the SURVIV trial. The study has a target enrolment of 200 patients with severe, symptomatic mitral bioprosthetic valve dysfunction (BVD) requiring reintervention, with an anticipated exclusion rate of approximately 50 patients who will either not meet the inclusion criteria, decline participation, or have other contraindications. The remaining 150 eligible patients will undergo 1:1 randomisation to either mViV (n=75) or surgical rMVR (n=75). Randomisation will be performed using a centralised, computer-generated allocation sequence with variable block sizes to ensure balanced treatment assignment and allocation concealment. All randomised patients will receive their allocated intervention according to standardised protocols, followed by comprehensive clinical assessments at predetermined intervals including 3 months, 12 months, and annually up to 10 years. The follow-up protocol encompasses clinical evaluation, multimodality imaging, and quality-of-life assessments designed to capture both acute safety outcomes and long-term efficacy endpoints. The primary analysis will follow the ITT principle, analysing all 150 randomised patients according to their assigned treatment group regardless of protocol deviations or crossover. CT: computed tomography; ITT: intention-to-treat; LVOT: left ventricular outflow tract; mITT: modified intention-to-treat; mViV: mitral valve-in-valve; rMVR: redo-mitral valve replacement; TOE: transoesophageal echocardiography; TR: tricuspid regurgitation; ViV: valve-in-valve
Eligibility and screening
Patients >18 years old with severe, symptomatic mitral BVD and requiring repeated mitral valve intervention will be screened for enrolment into the trial. Inclusion and exclusion criteria are depicted in Table 1. A complete medical history, physical examination, New York Heart Association (NYHA) Functional Class assessment, laboratory studies (complete blood count, metabolic panel, liver function tests, coagulation studies, B-type natriuretic peptide [BNP]/N-terminal [NT]-proBNP), and 12-lead electrocardiogram will be required. A comprehensive imaging assessment will be performed within 90 days prior to the procedure, including transthoracic and transoesophageal echocardiography (TOE) and cardiac computed tomography (CT). A coronary CT scan to exclude concomitant coronary artery disease (CAD) and/or a right heart cardiac catheterisation with invasive coronary angiography may be required to confirm pulmonary hypertension and assess associated CAD. As the available risk-stratification tools remain controversial in estimating outcomes after rMVR and mViV, Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) or European System for Cardiac Operative Risk Evaluation II (EuroSCORE II) cutoff values will be waived during the screening phase, yielding a pragmatic local Heart Team-centred decision regarding eligibility. A committee comprising investigators who are participants in the trial will review all screened cases to determine if the patient is an appropriate candidate for both treatments, with a focus on anatomical risk factors (e.g., valve sizing, risk of left ventricular outflow tract [LVOT] obstruction, porcelain aorta, hostile chest, and intracardiac thrombus) and any other relevant clinical factors that could impact enrolment eligibility (such as associated valvular or myocardial diseases). The risk of LVOT obstruction20 will be predicted using dedicated CT software (3mensio [Pie Medical Imaging]). Once deemed an appropriate candidate, the patient will be considered for enrolment in the study.
Table 1. Patient selection criteria.
| Inclusion criteria: |
|---|
| Age >18 years; Symptoms of heart failure NYHA Class >II; Severe mitral bioprosthetic dysfunction defined by echocardiography; The Heart Team agree on eligibility, including assessment that mViV and surgical rMVR are suitable; The study patient or the study patient’s legal representative is informed of the nature of the study, agrees to its provisions and provides written informed consent as approved by the institutional review board centre; The study patient agrees to comply with all required post-procedure follow-up visits including annual visits for 10 years; The Heart Team agree on treatment strategy for concomitant coronary disease (if present); The patient agrees to undergo surgical rMVR if randomised to control treatment. |
| Exclusion criteria: |
| Heart Team assessment of inoperability (including examining cardiac surgeon); Hostile chest; Acute myocardial infarction <1 month before the intended treatment (WHO definition); Concomitant, severe valvular disease (aortic, tricuspid or pulmonic) requiring surgical intervention; Mitral mechanical prosthesis or valve rings; Pre-existing mechanical or bioprosthetic valve dysfunction in another position; Complex CAD: unprotected LM, SYNTAX >32 (in the absence of prior revascularisation); Any therapeutic invasive cardiac procedure resulting in a permanent implant that was performed within 30 days of the index procedure (unless part of a planned strategy for treatment of concomitant CAD). Implantation of a permanent pacemaker is not an exclusion criterion; Patients with planned concomitant surgical or transcatheter ablation for atrial fibrillation; Leucopaenia (WBC <3,000 cells/mL), acute anaemia (Hb <9 g/dL), thrombocytopaenia (Plt <50,000 cells/mL); Hypertrophic cardiomyopathy with or without obstruction; Predicted neo-LVOT area <170 mm² without feasible augmentation strategies; Severe ventricular dysfunction with a left ventricular ejection fraction <20%; Echocardiographic evidence of a mobile intracardiac mass, thrombus, or vegetation; Active upper gastrointestinal bleeding within 3 months (90 days) prior to the procedure; A known contraindication or hypersensitivity to all anticoagulation regimens, or inability to be anticoagulated for the study procedure; Clinically or neuroimaging-confirmed stroke or TIA within 3 months prior to the procedure; Renal insufficiency (creatinine >3.0 mg/dL) and/or renal replacement therapy at screening; Estimated life expectancy <12 months due to active malignancies, severe hepatic dysfunction (Child-Pugh class C), severe COPD (FEV1 <30% predicted) or others; Currently participating in an investigational drug or another device study; Active bacterial endocarditis within 6 months of procedure; Current pregnancy or intention to become pregnant during the study period. |
| CAD: coronary artery disease; COPD: chronic obstructive pulmonary disease; FEV1: forced expiratory volume in 1 second; Hb: haemoglobin; LM: left main; LVOT: left ventricular outflow tract; mViV: mitral valve-in-valve; NYHA: New York Heart Association; Plt: platelets; rMVR: redo-mitral valve replacement; TIA: transient ischaemic attack; WBC: white blood count; WHO: World Health Organization |
Randomisation, treatment, and follow-up
After informed consent, patients will be randomised 1:1 to receive either transcatheter mViV with a SAPIEN 3 or a SAPIEN 3 Ultra THV platform (both Edwards Lifesciences) or rMVR with a surgical bioprosthetic valve. A subrandomisation in the surgical group will define which bioprosthetic valve is to be used – ensuring that one-third of surgical patients receive a PERIMOUNT or PERIMOUNT Magna Ease bioprosthesis (both Edwards Lifesciences) and two-thirds receive another commercially available surgical bioprosthesis. Randomisation will be performed using a centralised, computer-generated allocation sequence with variable block sizes to ensure balanced treatment assignment while maintaining allocation concealment. The electronic randomisation system will be accessible 24 hours a day through a secure web-based platform, ensuring rapid treatment allocation without delays that could compromise patient care. Randomisation will occur only after all eligibility criteria have been verified and informed consent has been obtained. The allocation sequence will be concealed from investigators until the moment of randomisation, preventing selection bias in patient enrolment. Given the nature of the interventions, blinding of patients and treating physicians is not feasible. However, efforts will be made to minimise bias through blinded endpoint adjudication, standardised imaging protocols, and objective outcome measures. The clinical events committee will remain blinded to treatment allocation when analysing primary and secondary outcomes at 1-year follow-up.
The assigned treatment will be performed following treatment guidelines and according to local best practices, as described in Supplementary Appendix 1. The choice of anticoagulant and the duration of the antithrombotic regimen after rMVR will be left to physician discretion. Nevertheless, a specific protocol will be advised, and for patients in sinus rhythm, anticoagulation with a vitamin K antagonist (warfarin) to achieve an international normalised ratio of 2.5, or alternatively a direct oral anticoagulant, will be recommended for at least 3 months after surgery. At the time that the study was designed, the evidence base for the optimal antithrombotic strategy after mViV was not robust. Hence, the same protocol for rMVR will also be recommended. Details regarding antithrombotic and antiplatelet therapy (including compliance and reasons for interruption) and other cardiac medications will be recorded at each study visit.
The follow-up protocol includes clinical evaluation, laboratory testing, imaging studies, and quality-of-life questionnaires (EQ-5D-5L) at predefined intervals and is outlined in Figure 2. Echocardiographic (TOE) and cardiac CT examinations at 3 and 12 months will be performed and independently assessed by an imaging core lab to validate findings and provide useful imaging data for prespecified substudies of bioprosthetic leaflet thrombosis and early signs of structural valve dysfunction. Echocardiographic variables include mean mitral transprosthetic gradients, mitral valve area, Doppler velocity index, assessment of central and/or paravalvular regurgitation, plus other standard evaluations; CT variables cover the predicted and true post-procedure neo-LVOT areas.
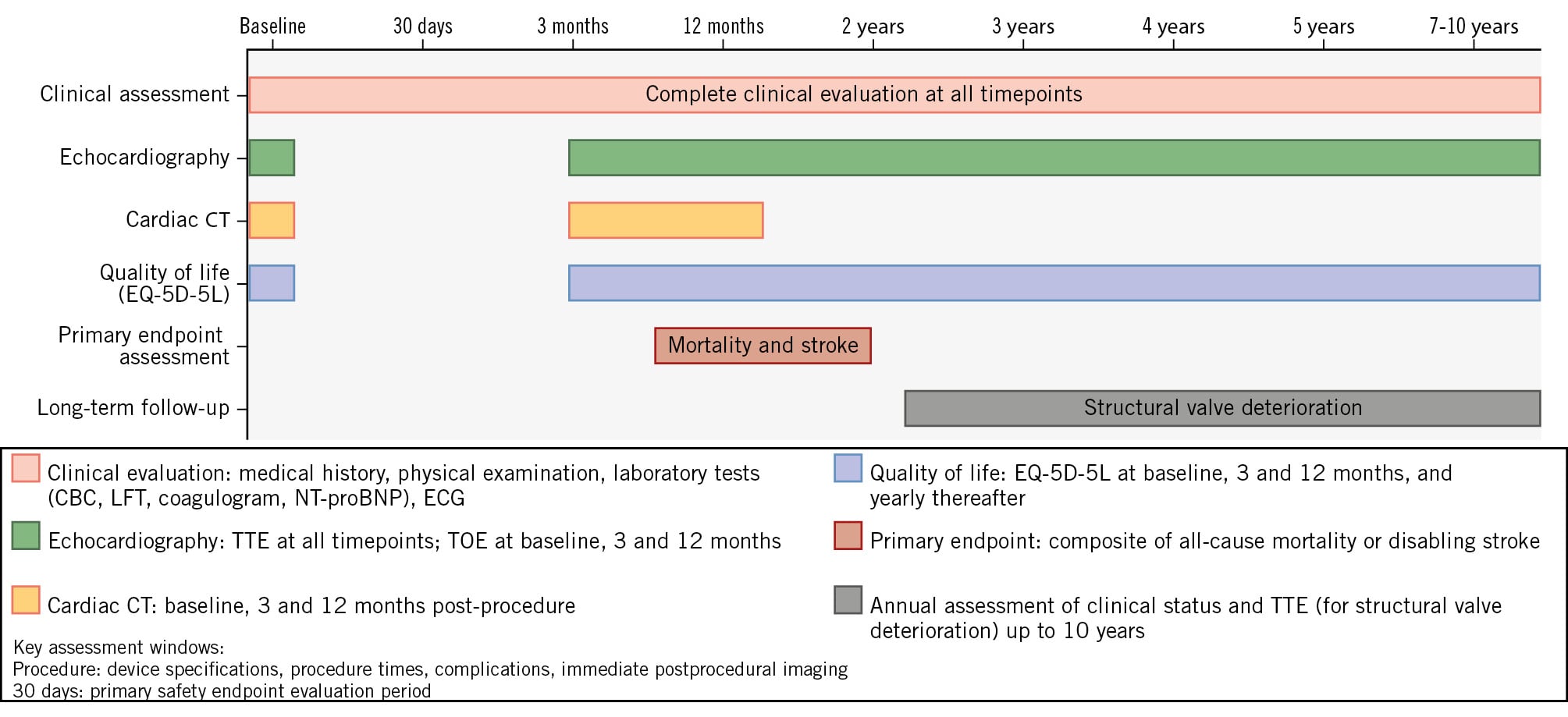
Figure 2. Study timeline and follow-up schedule. This comprehensive follow-up schedule illustrates the timing and scope of clinical assessments designed to capture both acute and long-term outcomes while minimising patient burden and ensuring high retention rates. Clinical assessment (pale red bar) includes a complete medical history, physical examination, laboratory studies (complete blood count, comprehensive metabolic panel, liver function tests, coagulation studies, and BNP/NT-proBNP), and 12-lead electrocardiography at all timepoints. Echocardiographic evaluation (green bars) consists of TTE at baseline, 3 months, 12 months, and annually thereafter, with TOE performed at baseline, 3 months, and 12 months for comprehensive anatomical assessment. Cardiac CT (orange bars) is performed at baseline, 3 months, and 12 months to evaluate anatomical suitability, for procedural planning, and to assess postprocedural complications (such as THV thrombosis or signs of early bioprosthetic deterioration). A quality-of-life assessment (blue bars) using the EQ-5D-5L questionnaire is administered at baseline, 3 months, and 12 months, and yearly thereafter to assess sustained functional improvements. The primary endpoint assessment (dark red bar) occurs at 12 months and evaluates the composite outcome of all-cause mortality or disabling stroke. Long-term follow-up (grey bar) continues annually from 2 to 10 years, focusing on structural valve deterioration, valve-related complications, and long-term durability outcomes. This structured approach ensures comprehensive data collection for both primary safety and efficacy endpoints while maintaining feasibility for long-term patient follow-up. BNP: B-type natriuretic peptide; CBC: complete blood count; CT: computed tomography; ECG: electrocardiogram; EQ-5D-5L: EuroQol 5-dimension 5-level; LFT: liver function test; NT-proBNP: N-terminal pro-B-type natriuretic peptide: THV: transcatheter heart valve; TOE: transoesophageal echocardiography; TTE: transthoracic echocardiography
Study endpoints
The primary and secondary endpoints are listed in Table 2. The primary endpoint is defined as the incidence of a composite of all-cause mortality or stroke (defined as disabling) at 12 months post-procedure. Secondary endpoints include safety outcomes at 30 days, efficacy outcomes at 12 months, and valve-related complications such as thrombosis and structural valve deterioration, assessed annually. Clinical outcomes will be defined in accordance with the Mitral Valve Academic Research Consortium21, as this was the most current consensus document at the time of the study’s design and first enrolment. Conversely, structural mitral bioprosthetic dysfunction and/or valve thrombosis will be classified according to newly proposed echocardiographic and cardiac CT definitions22.
Table 2. Primary and major secondary endpoints.
| Primary endpoint |
|---|
| All-cause death or disabling stroke at 12 months |
| Secondary endpoints |
| Safety endpoints at 30 days |
| All-cause mortality |
| Cardiovascular mortality |
| Disabling stroke |
| Major vascular complications |
| Life-threatening or major bleeding (BARC 3-5) |
| Acute kidney injury (AKIN stage 2-3) |
| New-onset atrial fibrillation |
| Reoperation for any cause |
| Left ventricular outflow tract obstruction |
| Efficacy endpoints at 12 months |
| Individual components of the primary endpoint |
| Cardiovascular death |
| Rehospitalisation for heart failure |
| Change in NYHA Functional Class |
| Valve-related complications (yearly) |
| Prosthetic valve thrombosis |
| Structural valve deterioration |
| Bioprosthetic valve failure |
| Endocarditis |
| Long-term endpoints (annually up to 10 years) |
| All-cause mortality |
| Cardiovascular mortality |
| Disabling stroke |
| Myocardial infarction |
| Heart failure hospitalisation |
| AKIN: Acute Kidney Injury Network; BARC: Bleeding Academic Research Consortium; NYHA: New York Heart Association |
Statistical analysis
Sample size calculation
The sample size calculation is based on the hypothesis of the superiority of transcatheter mViV implantation compared with conventional mitral valve surgery with respect to the 12-month composite primary endpoint. Available evidence comparing both treatments was minimal at the time of the protocol design in 2018. Therefore, the mortality rates were estimated from descriptive studies published between 2012 and 2017 – which represented the most contemporary data available at the time of protocol development. These studies report on the 1-year follow-up data for mViV or surgical rMVR separately2324252627. Moreover, stroke rates beyond 30 days were not reported systematically in the literature, so historical data from participating centres were used. Accordingly, we assumed an estimated cumulative event rate of 8% in the mViV group and 25% in the surgical rMVR group. Considering these proportions, a 2-sided significance level of 5%, and a statistical power of 80%, a total of 150 patients is estimated to be required. The calculation assumes a 1:1 randomisation ratio between groups and is based on a time-to-event analysis using a Cox proportional hazards regression model.
Statistical methods
Analyses will be conducted on the following two population sets. The intention-to-treat (ITT) set includes all randomised patients, analysed according to their originally assigned treatment groups, regardless of the actual treatment received or any protocol deviations.
The modified intention-to-treat (mITT) set includes all randomised patients, analysed according to their originally assigned treatment groups but considering clinical outcomes from the date of the procedure (if performed) rather than from the date of randomisation.
The primary analysis will use both the ITT and mITT population sets. The primary composite endpoint of all-cause mortality or disabling stroke will be analysed as a time-to-event outcome using Kaplan-Meier survival curves. Cox proportional hazards regression will be employed to estimate hazard ratios with 95% confidence intervals, adjusting for age. The proportional hazards assumption will be assessed using Schoenfeld residuals and graphical methods. If the assumption is violated, Greenwood-based Z-tests will be performed to compare the cumulated incidences at 3, 6, and 12 months.
Regarding secondary analyses, time-to-event endpoints will be analysed using Kaplan-Meier survival curves, and hazard ratios will be estimated using Cox proportional hazards models, adjusted for age. Proportional hazards assumption violations will be treated similarly to those in the primary endpoint analysis. Quality-of-life evaluation (EuroQol) will be compared between groups using linear regression models adjusted for age and baseline EuroQol values, and results will be presented as mean differences with 95% confidence intervals.
With respect to missing data, all time-to-event analyses will be censored at the last visit performed for each participant or at the time of death (if death is not the event of interest). No missing events will be imputed. Missing baseline or follow-up data will be handled using a complete case analysis. No imputation methods are prespecified.
In terms of additional analyses, we will calculate the restricted mean survival time at 1 year for the primary endpoint, and landmark analyses at 6 months will be presented to distinguish short- and long-term comparisons. Hazard ratios with 95% confidence intervals will be reported for each time window.
Continuous endpoints will be analysed using t-tests or Wilcoxon rank-sum tests, depending on data distribution. Categorical endpoints will be compared using the chi-square or Fisher’s exact test as appropriate.
Quality assurance
Statistical analysis will be conducted using R software, version 4.4.1 or later (R Foundation for Statistical Computing), with all analyses prespecified in a detailed statistical analysis plan that will be finalised before database lock. All statistical code will be documented and archived to ensure the reproducibility of results. Additional information regarding data collection and management is provided in Supplementary Appendix 2.
Clinical events committee
An independent clinical events committee comprising a cardiologist, an interventional cardiologist, and a cardiovascular surgeon will review and adjudicate all primary and secondary endpoint events using predefined criteria. Committee members will be blinded to treatment allocation and will base their assessments on comprehensive clinical documentation and imaging studies.
Data safety monitoring board
An independent data safety monitoring board (DSMB) will provide ongoing oversight of study conduct, safety, and efficacy. The DSMB will review unblinded safety data at regular intervals and have the authority to recommend study modification or termination if safety concerns arise.
Discussion
Bioprosthetic heart valves have been increasingly preferred over mechanical valves in patients who require surgical aortic or mitral valve replacement. When structural BVD ensues, surgical rMVR prevails as the treatment of choice, and studies have shown that – depending on patient age – a repeat surgical procedure can be required in up to 35% of patients at 10 years28 and 50% at 20 years29. Early postoperative mortality ranges from 6% to 12% in recent series345623242526, but this can be significantly higher in patients with multiple surgeries7, endocarditis, or pulmonary hypertension24.
The development of percutaneous treatment of aortic stenosis paved the way for a transcatheter approach for mitral BVD, and Cheung et al were the first to report a valve-in-valve procedure using transapical access30. Initial studies have demonstrated acceptable clinical and haemodynamic results, despite the high-risk characteristics of patients selected for these procedures27. A progressive shift towards a higher use of the transseptal approach has been observed, and recent evidence revealed lower mortality and stroke rates9. Analysing a large cohort of 4,243 high surgical risk patients (Society of Thoracic Surgeons score 9±8%) treated from 2015 to 2022, Goel et al10 found mortality rates of 4.3% at 30 days and 13.4% at 1 year, with a high technical success rate and a low incidence of periprocedural complications, such as LVOT obstruction (0.4%) or valve migration (0.3%). In a prospective, observational study with carefully selected patients (n=50) deemed at intermediate risk for surgical rMVR, there were no deaths or strokes at a median follow-up of 758 days, and the rate of technical success was 98.0%11. By avoiding sternotomy and cardiopulmonary bypass, mViV should trigger less systemic inflammation and tissue trauma, hasten recovery, and improve the clinical outcomes of patients with mitral SVD (Figure 3).
Several study-level meta-analyses revealed that – despite older age, more comorbidities, and a high surgical risk profile – patients undergoing mViV had a lower rate of periprocedural complications (including stroke, bleeding and acute kidney injury) and a shorter hospital length of stay12131415. Overall, no significant differences in mortality rates between rMVR and mViV were reported at 1 year, although a few studies revealed a lower in-hospital mortality rate after mViV1415. There are unsettled issues related to the mViV procedure, as it can result in smaller effective orifice areas, raising concerns about higher transprosthetic gradients and patient-prosthesis mismatch. The incidence of leaflet thrombosis has not been clearly documented, and late THV durability is largely unknown and needs to be addressed before expanding indications to low-risk patients. In this regard, rheumatic valve disease is still a common indication for cardiac surgery in younger adults, representing a challenge to public health in low- and middle-income countries; management of heart valve disease in these countries requires long-term follow-up and is characterised by the need for repeat operations, resulting in substantial impact on a patient’s quality of life and healthcare costs.
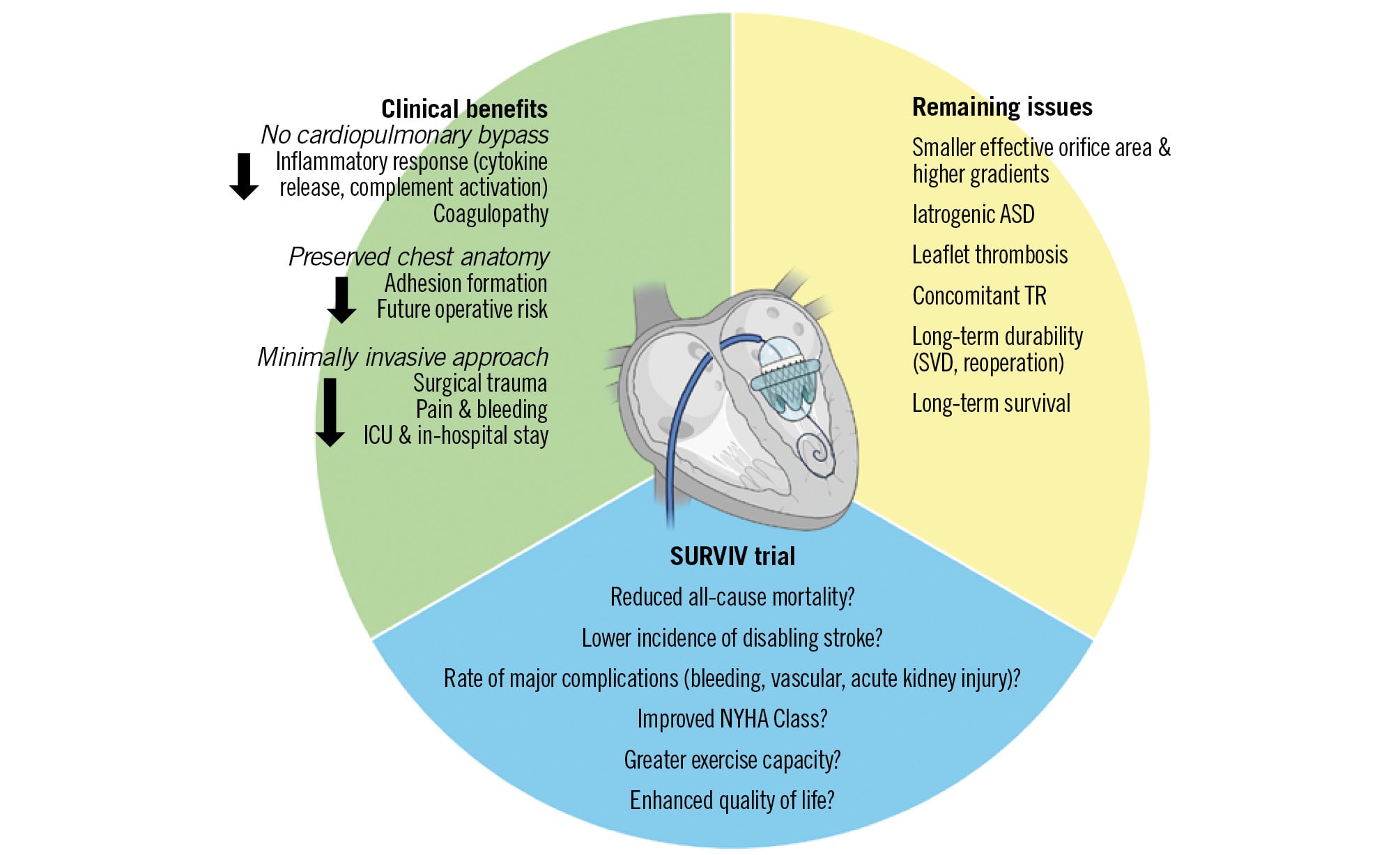
Figure 3. Mechanistic rationale and hypothesised clinical benefits of the transcatheter valve-in-valve approach. This conceptual framework illustrates the mechanistic rationale underlying the study hypothesis that transcatheter mitral ViV may demonstrate clinical benefits as compared to surgical rMVR for the 12-month composite primary endpoint. The transseptal ViV approach avoids sternotomy and cardiopulmonary bypass, theoretically providing key mechanistic advantages: reduced inflammatory response through decreased cytokine release and complement activation; elimination of cardiopulmonary bypass-related complications including coagulopathy; minimally invasive technique resulting in reduced surgical trauma, pain, and bleeding; and preservation of chest wall anatomy with decreased adhesion formation and lower future operative risk. These mechanistic benefits are hypothesised to accelerate patient recovery through faster mobilisation, shorter ICU stays, and earlier hospital discharge. The improved recovery profile is expected to translate into superior clinical outcomes across multiple domains: enhanced 30-day safety profile with reduced all-cause mortality, stroke incidence, life-threatening bleeding, and acute kidney injury; improved functional outcomes including better NYHA Functional Class, greater exercise capacity, enhanced quality-of-life scores, and superior symptom relief; and achievement of the primary composite endpoint of reduced all-cause mortality and stroke with sequelae at 12 months. Remaining issues related to transcatheter treatment that need to be addressed include the role of iatrogenic ASD and concomitant TR after the procedure and rates of leaflet thrombosis, SVD, reintervention, and long-term durability at 10 years as compared to surgical rMVR. ASD: atrial septal defect; ICU: intensive care unit; NYHA: New York Heart Association; rMVR: redo-mitral valve replacement; TR: tricuspid regurgitation; SVD: structural valve deterioration; ViV: valve-in-valve
Conclusions
SURVIV is the first randomised trial to compare transcatheter mitral valve-in-valve to redo-surgical valve replacement in patients with severe, symptomatic mitral bioprosthetic dysfunction. The results of this study could potentially provide evidence to impact treatment recommendations and support shared decisions for patients who need repeat mitral interventions.
Acknowledgements
We are indebted to Denise Abud, MD for her invaluable and highly professional support throughout the initial phase of development of this study.
Funding
The SURVIV trial is sponsored by the Instituto Dante Pazzanese de Cardiologia and Fundação Adib Jatene, São Paulo, Brazil. Additional support is provided by Edwards Lifesciences, including a research grant, device supply (75 transcatheter heart valves and 25 surgical heart valves) and technical assistance provided by clinical field specialists during transcatheter valve procedures. The Brazilian Unified Health System (SUS) is providing coverage for other treatment costs for all the enrolled patients. The funding sources had no role in the design of the study, collection and analysis of data, or preparation of this manuscript.
Conflict of interest statement
D.L. Bhatt discloses the following relationships - advisory board: AngioWave, Antlia Bioscience, Bayer, Boehringer Ingelheim, CellProthera, Cereno Scientific, E-Star Biotech, High Enroll, Janssen, Level Ex, McKinsey, Medscape Cardiology, Merck, NirvaMed, Novo Nordisk, Repair Biotechnologies, Stasys, SandboxAQ (stock options), and Tourmaline Bio; board of directors: American Heart Association New York City, AngioWave (stock options), Bristol-Myers Squibb (stock), DRS.LINQ (stock options), and High Enroll (stock); consultant: Alnylam, Altimmune, Broadview Ventures, Corcept Therapeutics, Corsera, GlaxoSmithKline, Hims, SERB, SFJ, Summa Therapeutics, and Worldwide Clinical Trials; data monitoring committees: Acesion Pharma, Assistance Publique-Hôpitaux de Paris, Baim Institute for Clinical Research, Boston Scientific (Chair, PEITHO trial), Cleveland Clinic, Contego Medical (Chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ABILITY-DM trial, funded by Concept Medical; for ALLAY-HF, funded by Alleviant Medical), Novartis, Population Health Research Institute, and Rutgers University (for the NIH-funded MINT Trial); honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Arnold and Porter law firm (work related to Sanofi/Bristol-Myers Squibb clopidogrel litigation), Baim Institute for Clinical Research (AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor-in-Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), CSL Behring (AHA lecture), Duke Clinical Research Institute, Engage Health Media, HMP Global (Editor-in-Chief, Journal of Invasive Cardiology), Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Oakstone CME (Course Director, Comprehensive Review of Interventional Cardiology), Philips (Becker’s Webinar on AI), Population Health Research Institute, WebMD (CME steering committees), and Wiley (steering committee); other: Clinical Cardiology (Deputy Editor); Progress in Cardiovascular Diseases (Deputy Editor); patent: Sotagliflozin (named on a patent for sotagliflozin assigned to Brigham and Women’s Hospital who assigned to Lexicon; neither he nor Brigham and Women’s Hospital receive any income from this patent); research funding: Abbott, Acesion Pharma, Afimmune, Alnylam, Amarin, Amgen, AstraZeneca, Atricure, Bayer, Boehringer Ingelheim, Boston Scientific, CellProthera, Cereno Scientific, Chiesi, Cleerly, CSL Behring, Faraday Pharmaceuticals, Fractyl, Idorsia, Janssen, Javelin, Lexicon, Lilly, Medtronic, Merck, MiRUS, Moderna, Novartis, Novo Nordisk, Pfizer, PhaseBio, Regeneron, Reid Hoffman Foundation, Roche, Sanofi, Stasys, and 89Bio; royalties: Elsevier (Editor, Braunwald’s Heart Disease); site co-investigator: Cleerly. D.A. Siqueira discloses the following relationships: proctor, Edwards Lifesciences. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
