Abstract
The treatment of coronary artery disease (CAD) in patients with severe aortic valve stenosis (AVS) eligible for transcatheter aortic valve implantation (TAVI) is not supported by clinical evidence, and the role of physiology over anatomy as well as the timing of coronary intervention are not defined. FAITAVI (ClinicalTrials.gov: NCT03360591) is a nationwide prospective, open-label, multicentre, randomised controlled study comparing the angiography-guided versus the physiology-guided coronary revascularisation strategy in patients with combined significant CAD and severe AVS undergoing TAVI. Significant CAD will be defined as coronary stenosis ≥50%, as assessed by visual estimation in vessels ≥2.5 mm. Physiology will be tested by fractional flow reserve (FFR) and instantaneous wave-free ratio (iFR). The study will be conducted at 15 sites in Italy. In the angiography arm, percutaneous coronary intervention (PCI) will be performed either before TAVI, during the TAVI procedure – before or after the valve implantation – or within 1 month±5 days of the valve implantation, left to the operator’s decision. In the physiology arm, FFR and iFR will be performed before TAVI, and PCI will be indicated for FFR ≤0.80, otherwise the intervention will be deferred. In case of borderline values (0.81-0.85), FFR and iFR will be repeated after TAVI, with PCI performed when needed. With a sample size of 320 patients, the study is powered to evaluate the primary endpoint (a composite of death, myocardial infarction, stroke, major bleeding, or ischaemia-driven target vessel revascularisation). TAVI indication, strategy and medical treatment will be the same in both groups. After discharge, patients will be contacted at 1, 6, 12 and 24 months after the procedure to assess their general clinical status, and at 12 months for the occurrence of events included in the primary and secondary endpoints. FAITAVI is the first randomised clinical trial to investigate “optimal” percutaneous coronary intervention associated with TAVI in patients with severe AVS and CAD.
The prevalence of coronary artery disease (CAD) ranges from 30% to 60% in patients with severe aortic valve stenosis (AVS)1. Observational studies reporting outcomes of patients undergoing transcatheter aortic valve implantation (TAVI) revealed a prevalence of CAD in the range of 40-75%2; these data are confirmed by the analysis of the SURTAVI and PARTNER IIA trials performed in intermediate-risk patients34.
While current guidelines5 state that myocardial revasculaÂrisation at the time of surgical aortic valve repair is a class I recommendation in the presence of coronary stenosis ≥70%, and a class IIa recommendation for angiographic stenosis 50-70%, the level of evidence (LoE) is poor (LoE C), and thus, the best management of CAD in TAVI candidates remains unclear. Current European Society of Cardiology (ESC) guidelines5 state that myocardial revascularisation using percutaneous coronary intervention (PCI) should be considered in patients with a primary indication to undergo TAVI and who present with coronary artery diameter stenosis >70% in proximal segments (class IIa, LoE C). The latest American Heart Association guidelines6 suggest that in patients presenting with concomitant severe AVS and significant CAD (luminal reduction >70% diameter, fractional flow reserve [FFR] <0.8, instantaneous wave-free ratio [iFR] <0.89) consisting of complex bifurcation left main and/or multivessel CAD with a Synergy Between Percutaneous Coronary Intervention With Taxus and Cardiac Surgery (SYNTAX) score >33, surgery is preferred over TAVI and PCI (class IIa, LoE C). A recent European Association of Percutaneous Cardiovascular Interventions (EAPCI) consensus gave similar recommendations on this topic7.
STUDY RATIONALE
Few studies have directly evaluated the impact of CAD on patient outcomes after TAVI, with conflicting results, highlighting that not all patients benefit from revascularisation before valve replacement and that the benefit of PCI in stable CAD might be limited to significant stenotic lesions in proximal coronaries only1. In previous studies, the presence of CAD was not associated with a worse outcome in the TAVI setting, but the definition of CAD was highly heterogeneous, including previous myocardial infarction (MI), previous coronary artery bypass grafting (CABG) or PCI, or coronary stenoses >50%8. A meta-analysis failed to find a benefit of PCI in the TAVI setting with respect to several patient-important clinical outcomes; on the contrary, it showed an increase in 30-day mortality9.
To date, the PercutAneous Coronary inTervention prior to transcatheter aortic VAlve implantaTION (ACTIVATION) study is the only randomised trial providing some insights on the topic. The study showed similar rates of death and rehospitalisation at 1 year between PCI and no-PCI strategies (based exclusively on angiographic assessment of the stenosis) prior to TAVI, with PCI resulting in a significantly higher incidence of bleeding10.
Considering that inducible myocardial ischaemia documented by intracoronary physiology may justify PCI in stable CAD patients with a low SYNTAX score1112, the hypothesis was made that the same method may prove beneficial in TAVI patients with incidentally found CAD by optimising the appropriateness of interventions, since previous studies have shown that the use of coronary physiology leads to a significant reduction of unnecessary PCI13.
Along these lines, the Nordic Aortic Valve Intervention (NOTION-3) Trial will answer the question of whether FFR-guided revascularisation improves outcomes compared to medical therapy alone in the presence of significant CAD, aimed at investigating the role of physiological assessment in the context of TAVI. However, in this trial, no comparisons with angiographic assessment are provided14.
Methods
STUDY DESIGN AND POPULATION
The Functional Assessment In TAVI (FAITAVI) study (ClinicalTrials.gov: NCT03360591) is an investigator-initiated, Italian nationwide, prospective, multicentre, randomised, open-label trial. The study is investigating the clinical outcome of patients presenting severe AVS and concomitant stable CAD treated by TAVI with a balloon-expandable SAPIEN 3/Ultra transcatheter heart valve (Edwards Lifesciences) and PCI according to 2 different strategies: 1) angiography-guided strategy versus 2) physiology-guided strategy. Details of the 2 strategies are reported in Supplementary Appendix 1. The study was approved and is coordinated by the Ethics Committee of the University of Verona (reference CESC1349). Figure 1 shows the trial design. A list of the FAITAVI investigators is included in Supplementary Appendix 2. FAITAVI eligibility criteria include age >18 years, severe symptomatic AVS with an indication for TAVI as proposed by the local Heart Team, and at least 1 coronary stenosis >50% per visual assessment in a major coronary artery (reference diameter >2.5 mm). The decision of the study group to select coronary stenosis >50% derives from our previous studies that demonstrated a different correlation between FFR and angiographic diameter stenosis in patients with severe AVS compared to patients without AVS, with FFR values becoming positive (0.80) with lower degrees of diameter stenoses1516.
A list of inclusion and exclusion criteria are presented in Table 1.
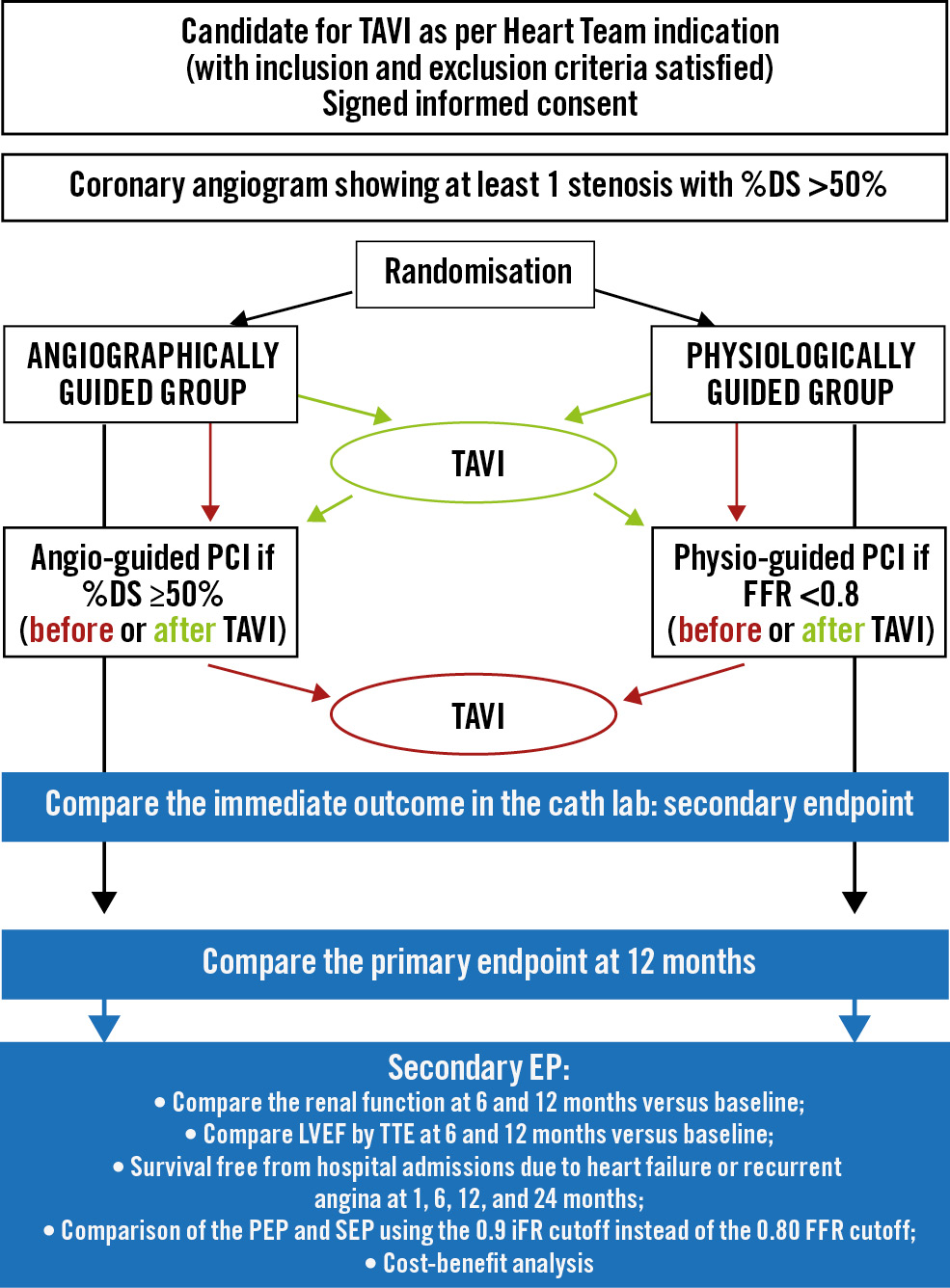
Figure 1. FAITAVI trial design. DS: diameter stenosis; EP: endpoint; FFR: fractional flow reserve; iFR: instantaneous wave-free ratio; LVEF: left ventricular ejection fraction; PCI: percutaneous coronary intervention; PEP: primary endpoint; SEP: secondary endpoint; TAVI: transcatheter aortic valve implantation; TTE: transthoracic echocardiography
Table 1. Key inclusion and exclusion criteria.
| Inclusion criteria | Exclusion criteria |
|---|---|
| Age >18 years | Pregnancy |
| Written informed consent | Left ventricular ejection fraction <35% |
| Diagnosis of severe native aortic valve disease with the indication to endovascular valve replacement given by Heart Team | Signs or symptoms of acute (unstable) myocardial ischaemia |
| Diagnosis of at least 1 coronary stenosis >50% at angiography | Contraindication to adenosine administration (e.g., asthma, chronic obstructive pulmonary disease, systolic blood pressure <90 mmHg) |
| Reduced survival expectancy due to severe comorbidities (<1 year) | |
| Impossibility of obtaining follow-up information | |
| Lack of any inclusion criteria |
SCREENING, ENROLMENT, AND RANDOMISATION PHASE
After verifying the selection (inclusion/exclusion) criteria, candidates will be first asked to sign the informed consent. Then, after coronary angiogram confirmation of the presence of significant CAD, consenting patients will be randomised to one of the 2 study arms according to an automatic centralised allocation system accessible 24 hours a day.
Study data will be collected on a dedicated electronic case report form (e-CRF) and managed using REDCap (Research Electronic Data Capture) tools17 hosted at the University of Padua, Padua, Italy.
PROCEDURAL DETAILS
GROUP 1: ANGIOGRAPHY-GUIDED REVASCULARISATION
Patients allocated to this group will undergo PCI and stenting of all coronary stenoses >50%, as assessed by visual estimation, in vessels with a diameter ≥2.5 mm. PCI can be performed before TAVI (preventive revascularisation), during the TAVI procedure – before or after the valve implantation – or within 1 month±5 days of the valve implantation. Implantation of a second-generation drug-eluting stent (DES) is advised in all interventions, and the brand of the stent is left to the operator’s choice. All major epicardial coronary arteries can be treated, including the left main trunk, with no limit on the number of stents implanted. In the presence of tandem lesions, both should be treated.
GROUP 2: PHYSIOLOGY-GUIDED REVASCULARISATION
Patients randomised to this group will undergo stenting of coronary lesions showing FFR values ≤0.80 only18. FFR and iFR measurements will be obtained in all the analysed lesions according to the operator’s evaluation (≥50%). Hyperaemia will be obtained after administering intracoronary boluses of 150 mg to 300 mg adenosine, as previously indicated19. This strategy has already been reported as reliable, reproducible and with diagnostic capabilities equivalent to intravenous adenosine infusion20. Nitroglycerine administration is not recommended before valve replacement21.
Haemodynamic parameters, including systemic blood pressure, heart rate, central venous pressure, left ventricular end-diastolic pressure, and peak-to-peak gradient, will be recorded both before and after TAVI, in order to assess haemodynamic conditions that could potentially alter the FFR and iFR measurements.
Physiological measurements will firstly be obtained before valve implantation in all cases using standard techniques, as detailed in dedicated studies111222. iFR measurements should always be obtained in each lesion before FFR measurements to avoid the effects of residual adenosine-induced hyperaemia and to minimise the procedural time.
Physiology assessment will guide the PCI strategy as follows:
• Lesions showing positive FFR measurements (0.80) must be treated with PCI, either before or after TAVI, according to the operator’s preference23.
• Lesions showing clearly negative values (FFR>0.85) will not be treated with PCI before TAVI, and repeated FFR and iFR measurements after TAVI are recommended.
• Lesions showing “borderline” FFR measurements before TAVI (FFR 0.81-0.85) should be reassessed after TAVI, and the decision of treating or deferring treatment will be based on the FFR value obtained after TAVI, as previously demonstrated22.
• In all cases, iFR values will be recorded for a post hoc analysis and for validation of the study endpoints according to iFR values.
• In the presence of tandem lesions with a positive distal FFR value, the proximal (most important) lesion will be treated first, and a new FFR measurement will determine whether or not to treat the most distal stenosis.
All physiological and angiographic measurements will be evaluated and adjudicated by a centralised laboratory analysis.
FUNCTIONAL ASSESSMENT FOLLOW-UP SUBSTUDY
For the physiology-guided group, in case of deferred treatment, a repeated measurement of iFR and FFR in the same vessel can be performed a few months after the index procedure, when haemodynamic changes induced by the valve replacement might have occurred.
Previous studies have shown that FFR remains quite stable during 6 months of follow-up, which is different from resting indices such as iFR and resting full-cycle ratio (RFR)2425.
The follow-up coronary angiography and iFR-FFR measurements are expected in a time window of 6 to 12 months after the valve implantation. Eligibility for follow-up angiography will be considered according to patients’ procedural risk and left to the investigators’ decision.
TRANSCATHETER AORTIC VALVE IMPLANTATION
Implantation of the transcatheter valve will be limited to the SAPIEN 3 and SAPIEN 3 Ultra valve models, and only patients treated through the transfemoral approach will be included. According to each patient’s characteristics and local protocols, the procedure will be performed either under general anaesthesia or local anaesthesia with mild sedation and by either percutaneous or surgical vascular access.
MEDICAL THERAPY
The concomitant therapy administered before, during and after the TAVI follows the latest evidence-based indications available at the time of the index procedure therapy5626, as follows:
• Before TAVI, all patients will be preloaded with a loading dose of clopidogrel and aspirin.
• Patients will receive appropriate anticoagulation and other therapy according to standard hospital practice to maintain an activated clotting time >280 seconds during the procedure. Either unfractionated heparin or bivalirudin may be used for procedural anticoagulation. Reversal of anticoagulation with protamine is recommended at the end of the procedure if it is not contraindicated by a high thrombotic risk.
• After successful valve implantation, patients receive single/ÂÂdouble antiplatelet therapy or anticoagulation in different combinations as appropriate, according to bleeding risk and/or PCI performance, as recommended by guidelines101827.
• Patients will be informed of the prescribed antiÂthrombotic treatment after the procedure, and the importance of treatment compliance throughout the study will be underlined to the patient. The details about antiÂplatelet therapy (including start and stop times of interrupted therapy and reason for interruption) and other cardiac medications will be recorded at each study visit. All other medication will be given at the discretion of the physician in charge, as clinically indicated.
FOLLOW-UP
Clinical follow-up at the outpatient clinic or using telemedicine will be performed at 1, 6, 12 and 24 months after the TAVI procedure. The visit report will include information regarding symptoms, vital signs, clinical information regarding the primary and secondary endpoints, according to the Valve Academic Research Consortium (VARC)-228 and Bleeding Academic Research Consortium (BARC)29 statements, together with the need for target vessel revascularisation (TVR) and any hospitalisation due to heart failure or myocardial ischaemia within 12 months of the index procedure.
Drug regimens, compliance with medical therapy, other treatments, and the need for hospitalisation due to extraÂcardiac reasons will also be monitored. All information will be verified by a primary care physician and/or hospital charts and death certificates. Follow-up angiography will be performed in patients complaining of suspected ischaemic symptoms within 1 year after the procedure to exclude possible restenosis or vessel occlusions and in those participating in the functional follow-up substudy.
STUDY ENDPOINTS
The primary endpoint is defined as the incidence of the composite occurrence of death (from any cause), myocardial infarction (both periprocedural and spontaneous), stroke (intended as disabling), major bleeding (as classified by the BARC29), or ischaemia-driven TVR (ID-TVR).
The main secondary, statistically powered endpoint is the comparison of the immediate outcome of the procedure (safety endpoint), assessing the possible relation between the presence of a significant CAD and the TAVI procedure as evaluated by the VARC-2 “early safety” endpoint, i.e., the composite endpoint of all-cause mortality, all strokes (disabling and non-disabling), life-threatening bleeding, acute kidney injury (stages 2 or 3, including the need for dialysis), coronary artery obstruction requiring intervention, major vascular complication, or valve-related dysfunction requiring a repeat procedure (aortic balloon valvuloplasty, TAVI, surgical aortic valve replacement) within 30 days.
The complete list of secondary endpoints and endpoint definitions are reported in Supplementary Appendix 3.
STATISTICAL CONSIDERATIONS AND SAMPLE SIZE CALCULATION
Available literature focusing on this specific issue was minimal at the time of the protocol design in 2016. Therefore, the event rates were estimated, based on previous studies reporting on the 1-year follow-up in CAD patients after TAVI30, real-world, long-term outcome reports in TAVI patients31, the original FAME study11, and a prospective TAVI registry performed at the University of Verona showing that about 50% of AVS patients present with concomitant CAD (Verona Valve Registry, CESC1918), with data published thereafter within an observational study on the same topic32.
The study is designed to show the superiority of physiology-Âguided revascularisation over angiography-guided revascularisation.
The primary study outcome is a composite occurrence of death (from any cause), myocardial infarction (both procedure related and spontaneous), stroke (intended as disabling), major bleeding (as classified by VARC-2) or need for TVR within 12 months of the index procedure. This time-to-event endpoint will be compared in the treatment groups (angiography-Âguided vs functionally guided PCI) using a proporÂtional hazards model.
The study design is defined as accounting for a 1-year risk of the composite endpoint (Ï) of about 32.5% in the angiography-guided group, and a 20.5% risk in the physiology-guided revascularisation group, with a higher bleeding risk in the first follow-up period33. Thus, a non-Âproportional accumulation or risk of the event has been incorporated in the power calculation as a Weibull risk function with a shape parameter equal to 0.5 and a scale parameter equal to 0.024. An equally balanced 1:1 randomisation ratio has been assumed between sample sizes. Based on the expected incidence rates in the experimental and control groups, the median event-free survival time (m), estimated at 12 months for both groups, has been calculated with the inverse of the Brookmeyer-Crowley formula34:
m = 12 log (0.5) / log (1 − Ï)
This resulted in a median event-free survival time of 21 months for the angiography-guided group and about 36 months for the physiology-guided group, corresponding to an approximate hazard ratio (HR) of 1.71. A 12-month fixed-time accrual period has been assumed, followed by a 12-month fixed-time follow-up.
As an interim analysis is foreseen at 6 months, we kept the alpha level at 0.025 to allow control of the overall error rate at the end of follow-up, which should be a maximum of 0.05. A power of 0.80 was assumed.
Based on these calculations, a total of 276 patients (138 per arm) was calculated as the number necessary for inclusion. However, to account for a possible 7% dropout rate, the sample size n will be increased to a total of 320 patients by considering the formula35 N=n/(1-R)2=320.
The hazard comparison across groups will be performed using a Cox regression model estimate. The 95% confidence interval estimates will also be reported. The proportionality of hazard will be assessed by visual inspection of the Schoenfeld residuals plot.
The secondary time-to-event endpoints will also be analysed with a Cox model.
The renal function and the left ventricular ejection fraction progression will be analysed using linear mixed-effect models to consider the correlation within repeated measurements at different timepoints in the study.
An alpha level of 0.05 will be used to define a statistically significant effect. A sample size estimation was performed using the R system36 and the NPHMC (Sample Size Calculation for the Proportional Hazards Mixture Cure Model; both R Foundation for Statistical Computing) libraries37. Analyses will also be performed using the R system.
ETHICAL CONSIDERATIONS, DATA MANAGEMENT AND PERSONAL DATA PROTECTION
The ethics committees and/or competent authorities, as required per applicable local regulations, will approve the study before any patient enrolment. The study will comply with the Declaration of Helsinki and ISO 14155. Subject data will be managed following the European Union’s General Data Protection Regulation (EU) 2016/679.
Discussion
STUDY ORGANISATION AND TIMELINES
FAITAVI is an investigator-initiated trial funded by Edwards Lifesciences and Philips/Volcano Corporation. A steering committee has been established to assist the study sponsor with designing and managing the study, overseeing its scientific validity, the quality and integrity of the data, and the dissemination of the study results through appropriate scientific presentations and publications.
After reviewing the original source documents, all events will be revised and adjudicated periodically by a clinical events committee, blinded to treatment assignment (Supplementary Appendix 4). The angiographic severity of CAD, along with the quality of functional evaluations, will be analysed in a dedicated laboratory, headquartered at the University Hospital of Verona, Verona, Italy (Supplementary Appendix 5). The independent Data Monitoring Committee (DMC; based at the Service for Clinical Trials and Biometrics, Unit of Biostatistics, Epidemiology and Public Health, University of Padua, Padua, Italy) will monitor the safety and well-being of the participating subjects, ensure the study’s scientific integrity, and recommend actions based on potential safety issues, including study suspension or termination based on prespecified criteria.
Investigators will be required to report any serious adverse events and unanticipated problems occurring during the study period to the DMC and local ethics committees as soon as they become aware of them. Interim analyses are planned to rule out any safety issue related to the study design.
The steering committee and the sponsor will perform interim safety and final statistical analyses according to the statistical analysis plan. Study enrolment occurred between November 2017 and June 2023, with a delay of 3 years due to the COVID-19 pandemic and related healthcare service’s needs. The primary endpoint results are anticipated in the third quarter of 2024.
Conclusions
The FAITAVI trial is the first randomised clinical trial investigating the “optimal” percutaneous revascularisation strategy associated with TAVI, using balloon-expandable SAPIEN 3/Ultra transcatheter heart valves, in patients with severe AVS and stable CAD.
Acknowledgements
We wish to thank the members of the Clinical Events Committee, the Service for Clinical Trials and Biometrics of Padua, and the centralised Laboratory of Verona for their precious help and support during the course of the study. We would also like to thank Prof. Carlo Di Mario from the University of Florence and Dr Francesco Bedogni from Ospedale San Donato Milanese for their willingness to participate in the study, without consideration for budget constraints.
Funding
The study is funded by an unrestricted grant provided by Edwards Lifesciences and Philips/Volcano Corporation. The funders have no role in the study design; data collection, management, analysis and interpretation; writing of the report; or the decision to submit the results.
Conflict of interest statement
F. Ribichini has received honoraria from and is proctor for Edwards Lifesciences and Medtronic. G. Tarantini has received lecture fees from Medtronic, Edwards Lifesciences, Abbott, and Boston Scientific. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
